The function igraph_to_phylo takes a taxon graph (see
get_taxon_graph) and attempts to convert that to a
a phylo object from ape.
This will fail if the graph is not simple (no loops or multiple edges)
or is not connected (has isolated taxa). Neither of these conditions
should happen in datasets but they do.
igraph_to_phylo(g)
get_phylogeny(record)Arguments
- g
A taxon graph returned by
get_taxon_graph- record
A single dataset record id
Value
An phylo object.
Details
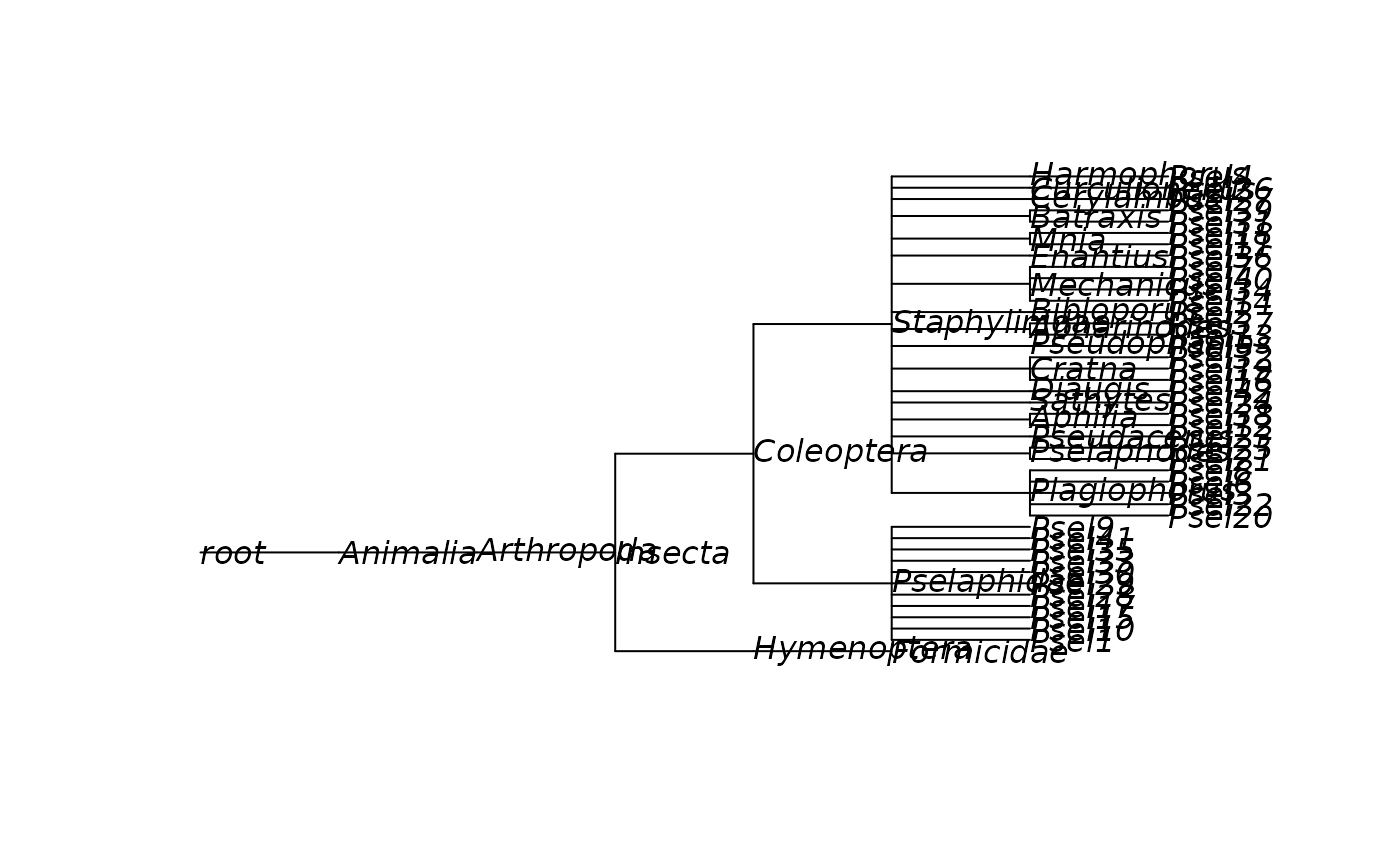
The phylogeny is assigned with equal branch lengths and so displays
showing the taxonomic hierarchy of taxa. Note that the phylogeny will
contain singleton nodes if an internal taxon has a single descendant -
see the example below showing internal node labels. The
ape functions
has.singles and
collapse.singles can be used to detect and remove
these if required.
The function get_phylogeny is simply a wrapper that calls
get_taxon_graph and then igraph_to_phylo.
Functions
get_phylogeny: Get a phylogeny for a dataset
See also
Examples
set_example_safe_dir()
#> Loading and caching index
#> Validating directory
beetle_graph <- get_taxon_graph(1400562)
beetle_phylo <- igraph_to_phylo(beetle_graph)
ape::plot.phylo(beetle_phylo, show.node.label = TRUE)
# Or wrapped into a single function
beetle_phylo <- get_phylogeny(1400562)
ape::plot.phylo(beetle_phylo, show.node.label = TRUE)
 unset_example_safe_dir()
#> Loading and caching index
#> Validating directory
unset_example_safe_dir()
#> Loading and caching index
#> Validating directory